| Оствальд был среди тех европейски ученых, которые открыли и оценили работы Гиббса, В 1892 году он перевел статьи Гиббса по термодинамики на немецкий язык. Катализ является одним из основных направлений физической химии и в частности термодинамики. Рассмотрим сначала очень кратко основные этапы развития физической химии. Их можно выделить четыре:
1. Подготовительный период, Методы математической обработки количественных анализов.
начиная с момента применения физических приборов для изучения физико-химических явлений до 40-х годов XIX века, когда был открыт закон сохранения и превращения энергии.
2. С 40-х до конца 70-х годов XIX века формирование отдельных направлений физической химии: электрохимии, термохимии, учение о равновесии, а так же становление первых научных основ физической химии.
3. С конца 70-х до 1913 года — взлет творческой физико-химической мысли. Постепенная ее эволюция перешла в 80-90-е годы XIX века в бурную революцию.
4. Примерно с 1913 года и до наших дней, ,боровская теория атомов.
Уже в подготовительный этап развития физической химии начали появляться догадки о существовании катализаторов и катализа. Бурное развитие катализа наблюдалось в последующие два этапа. Во второй половине XIX века в связи с успешным развитием органической химии, учения о катализе как бы замерзли на долгое время, несмотря на то, что осуществлению некоторых синтетических проблем могло произойти только благодаря катализу, но на это мало обращали внимания, и механизм катализа упускался из виду. Только в начале XX столетия катализ вновь обращает на себя внимание, Итак, рассмотрим истоки возникновения катализа.
Истоки возникновения учения о катализе
Еще в книге «Пиротехния»; опубликованной Бирингуччо в 1540 г., отмечалась роль времени в химических превращениях, В дальнейшем В. Гомбрег в 1700 г.. В, Льюис в 1759 г. и К. Венцель в 1777 г. вновь возвращались к этому вопросу. Особенно интересна работа немецкого химика Венцеля, в которой он высказал основные положения закона действия масс, сформулированного в более общем виде спустя почти столетие норвежскими учеными К, Гульдбергом и П. Вааге. Оствальд говорил о периоде развития химии после работ Венцеля до 50-60-х годов XIX в: «После полного надежд начала ... наступает очень длительная пауза, только один раз прерванная отдельными замечаниями теоретического характера. В «Химической статике» Бертолле упоминается о медленных химических процессах, как о явлениях «распространения» химических реакций. Бертолле, так же как и Венцель, является одним из предшественников, а не родоначальником химической кинетики.
Только во второй половине XIX века были созданы условия для плодотворного изучения скоростей реакций (проверка предполагаемых механизмов их протекания). Очень большое значение при создании предпосылок для возникновения химической кинетики сыграло изучение катализа.
Уже в работах К. Кирхгофа, Л, Тенара, X, Дэви и И. Деберейнера, опубликованных в 1912-1925 года, было описано множество каталитических реакций. Большая часть первых гипотез катализа ограничивало каталитическое влияния агента рамками только физического воздействия на ход реакций, Эти гипотезы подчеркивали, что любое химическое изменение реагентов протекает по законам стехиометрии.
Несколько иной путь рассмотрения каталитических реакций предложил шведский химик Берцелиус в 1835 году, открывший в химии эпоху катализа. По его мнению, «каталитическая способность (каталитическая активность в современном понимании) многих как простых, так и сложных тел в твердом виде и форме раствора является одним из проявлений электрохимических отношений материи. Большой заслугой Берцелиуса для выработки предпосылок возникновения кинетики и для ее становления явилось то, что он сумел обобщить проведенные ранее исследования вне стехиометрического влияния веществ на различные реакции и объединить все эти процессы общностью причины каталитической силы, Берцелиус называл каталитическую силу «причиной химического действия» и выдвинул тезис о «тысячи каталитических реакций в организме». Хотя Берцелиус не смог найти единого объяснения механизмов «каталитических процессов», содержащиеся в его работе обобщения (отграничение каталитических процессов, а также показ широкой распространенности и специфики каталитических превращений), способствовали более деятельному ограничению механизмов каталитических реакций.
Впервые объяснение катализа с единой точки зрения дал Либих в 1839 году, который считал, что каталитические явления обусловлены нарушением равновесия в притяжении радикала к элементам или радикала с которыми он связан вследствие роста химической разницы составляющих его элементов.
Это нарушение вызывается;
1. Изменение состояния сцепления, которые два или более элементов испытывают под влиянием теплоты;
2. Соприкосновением с третьим телом, которое при этом не входит в соединение;
3. Прибавлением элементов воды;
4. Одновременным совместным действием этих причин.
Таким образом, Либих связывал отклонение от стехиометрии с непрерывностью химического взаимодействия, начатого под действием этих причин, считая, что катализатор при этом остается химически неизменным. Теория Либиха явилась обобщением формальных физических (действие химически неизменного катализатора) и химических (стехиометрических) представлений о механизме каталитических превращений,
Развитие идей Либиха в XIX веке было скорее умозрительным совершенствованием гениальных предвидений, чем созданием рабочего инструмента для исследования природы катализа, основанного на реально существовавшем уровне знаний и экспериментальных возможностей химиков.
Интересные представления развивал Р. Майер, который рассматривал катализ как частный случай разряжения, Разряжение представляет «стековой курок», приводящий в действие большие количества «дремавшей» энергии.
Концепция Майера в применении к катализа получила развитие у А. Рея и в особенности у Оствальда.
В последней четверти XIX века Германия занимала ведущее положение в области исследований физических изменений, связанных с химическими реакциями,
Оствальд был среди тех европейски ученых, которые открыли и оценили работы Гиббса, В 1892 году он перевел статьи Гиббса по термодинамики на немецкий язык. Оствальд практически сразу начал применять теории Гиббса при изучении катализа. Катализ (термин, предложенный Берцелиусом в 1835 году) — изменение скорости химической реакции в присутствии небольших количеств веществ (катализаторов), которые не принимают видимого участия в реакции. Так, в 1816 году Дэви установил, что порошкообразная платина во много раз ускоряет присоединение водорода к кислороду и различным органическим соединениям. А Кирхгоф показал, что кислота значительно ускоряет расщепление ряда органических соединений. Причем ни платина, ни кислота в процессе реакции не расходуются, количество их остается неизменным.
Оствальд указал, что теория Гиббса заставляет предположить, что катализаторы, ускоряют реакции, не вызывая изменения в соотношении энергий взаимодействующих частиц, Катализатор, утверждал Оствальд, образует с исходным веществом промежуточное соединение, которое распадается на конечные продукты реакции, При распаде промежуточного соединения катализатор высвобождается. В отсутствии катализатора, то есть в отсутствии образуемого катализатором промежуточного соединения, данная реакция протекает намного медленнее, возможно даже практически незаметно. Таким образом, катализатор ускоряет реакцию, но сам при этом не расходуется. Кроме того, поскольку молекулы катализатора используются снова и снова, для ускорения реакции большого количества веществ достаточного небольшого количества катализатора.
Этот взгляд на катализ сохраняется и сегодня. Он помог объяснить механизм действия белковых катализаторов (или ферментов) управляющих химическими реакциями в организме.
Таким образом, в XIX столетии можно выделить следующие основные концепции по вопросу о роли катализа в химическом превращении. Катализатор — причина химической реакции, и его присутствие — непременное условие ее протекания (по Бсрцелиусу); нарушение равновесия в притяжении радикала к элементам или радикала (по Либиху). с которыми он связан; катализатор играет роль спускового курка (по Майеру) и наконец, катализатор ускоряет химическую реакцию, которая идет сама по себе, то есть катализатор управляет, но не движет химическим превращением (по Оствальду).
Принципиально новый этап в развитии учения о катализе начался после создания химической кинетики, когда разработка проблем катализа была переведена на «рельсы» кинетики химических реакций.
Как отмечал в 1909 году Оствальд «до создания учения о скорости химических реакций не представлялось возможности вывести целесообразные понятия относительно катализа, так как последний состоит в изменении скорости химических реакции вследствие присутствия веществ, отсутствующий в конечных продуктах реакции».
Уже Берцелиус и Либих упоминали об использовании времени превращения для характеристики активности катализаторов. Однако закон скоростей реакций был сформулирован Л. Вильгельми в 1850г. при исследовании проникании инверсии тростникового сахара, катализируемой неорганическими кислотами (HCl, HNO3, H2SO4 и Н3РO4):
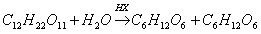
левулеаза декстроза
В приведенной формуле, «характеризующей химический процесс», скорость реакции пропорциональна действующим количествам сахара и катализирующей кислоты:

где:
dZ/dt - изменение количества сахара dZ за время dt (скорость процесса),
M - среднее количество сахара, которое подвергалось инверсии в течение бесконечно малого промежутка времени dt под действием каждой имеющейся единицы концентрации катализирующей кислоты (константа скорости реакции),
Z и S - соответственно количество сахара и катализирующей кислоты.
Следует упомянуть немецких химиков И. Левенталя и Е, Ленссена, которые в 1862 году независимо от работ Вильгельми сравнили действие различных кислот на протекание каталитической инверсии тростникового сахара
В 1882 году Меншуткин обнаружил ускорение химической реакции ее продуктами, то есть показал наглядный пример автокатализа,
Начиная с этого же года, Оствальд предпринял ряд исследований относительно каталитической активности кислот некоторых гидролитических реакций (гидролиз ацетамида, омыление метилацетета).
Таким образом, первыми объектами были каталитические реакции органических соединений, а так же реакции окисления, Становление кинетики химических реакций завершает классическая работа Я.Г. Вант-Гаффа «Очерки по химической динамике» (1884г.). С этого момента кинетика превращается из метода исследования в раздел физической химии. С утверждением кинетики химических реакций связан принципиально новый подход к изучению проблем катализа.
Наряду с чисто кинетическим изучением строения органических соединений в 80-90-х годах XIX века физико-химики искали связь между электрохимическим строением и реакционной способностью различных молекул. Это направление исследований получило значительное развитие после открытия в 1884 году Аррениусом и Оствальдом пропорциональности между скоростью каталитических жидкофазных реакций и электропроводностью растворов катализаторов.
Явления, наблюдаемые исследователями при изучении влияния протекания реакций, способствовали возникновению новых представлений о катализе, которые наиболее четко сформулировал Оствальд в 80-90-х годах XIX века.
С 1884 года Оствальд начал детальное изучение каталитического действия различных кислот на течение гидролитических реакций. При этом он впервые связал причину кислотного катализа с влиянием протона как основной часть кислот типа НХ. После появления теории электролитической диссоциации взгляды Оствальда на механизм кислотного катализа трансформировались в теорию кислотно-основного катализа, согласно которой ионы H+ и гидроксила являются наиболее эффективными катализаторами.
В гамбургском докладе 1901 года Оствальд сделал важнейший вывод о сущности катализа: катализ — это способ ускорения естественно протекающих реакций посредством введения в реакционную систему специальных посторонних веществ — катализаторов».
Исследование Оствалъда кинетики оказало очень большое влияние на развитие учения о катализе. Оствальд считал, что катализом является ускорение реакции, а не ее инициирование. Изучение кинетики каталитических превращений с привлечением кинетики газов создало к концу XIX века предпосылки создания фундаментального понятия химической кинетики энергии активации.
Развитие теоретических представлений о механизмах каталитических реакций.
Выдвинутые в 20-30-х годах представления о механизмах каталитических реакций в большинстве своем опирались на положения кинетических теорий. Однако, поскольку сфера применения каталитических теорий была уже, .чем общекинетических, задачи перед каталитическими концепциями стояли более прагматические — они должны были хорошо объяснить механизмы конкретных каталитических превращений, позволить делать прогнозы о протекании аналогичных реакций и дать возможность предсказывать новые катализаторы. Этим и вызван более локальный, «приземленный» и в большинстве случаев вторичный характер каталитических представлений в 20-30-х годах XX века по сравнению с созданными в то же время основными кинетическими теориями.
В связи с внедрением в химию электронных представлений в 1910 году, а также в связи с интенсивным развитием в это время экспериментальных методов исследования каталитических превращений, теории катализа, созданные в 20-30-х годах XX века, гораздо более детально рассматривают механизм взаимодействия в системе катализатор – реагент, чем изложенные ранее каталитические теории XIX — начала XX веков.
Рассмотрим основные теории катализа, созданные в 20-30-х годах XX века.
Теории промежуточной хемосорбции.
В начале 30-х годов были выдвинуты интересные гипотезы о связи между непрерывности изменения химического сродства и катализатором, опиравшиеся на достижения гетерогенного катализа, В работах И. Ленгмюра, А.А, Баландина, X. Тейлора и некоторых других химиков было показано, что первичный акт гетерогенного катализа представляет собой валентное взаимодействие атомов или молекул реагента с поверхностными атомами катализатора, то есть формой взаимодействия катализатора с реагентами является промежуточная хемосорбция. В этой теории произошел формальный синтез физических (роль адсорбции в катализе) и химических (валентное взаимодействие катализатора с реагентом) концепции катализа.
Дальнейшим развитием общей концепции промежуточной хемосорбции явились теории, конкретизирующие представления о поверхностных соединениях и о механизме их образования. Это послужило поводом к систематическому исследованию роли структуры поверхности в катализаторе, что нашло отражение в теории активных центров и статической теории активной поверхности.
Изучение хемосорбции привело далее к созданию теории активированной адсорбции.
Более высокий уровень в развитии концепции промежуточной хемосорбции был достигнут в конце 1920 — начале 1930-х годов при создании первой из общих теорий, связывающих образование поверхностных соединений с инициированием реакций. управлением активностью твердых катализаторов и селективностью их действия — мультиплетной теории (А. А. Баландин, 1929-1930 годах).
Мультиплетная теория катализа (теория промежуточных образований)
Согласно представлениям Баландина, при каталитическом превращении происходит расслабление и последующее перераспределение связей между реагирующими атомами, временно связанными с несколькими атомами катализатора в мультиплетном комплексе. При этом механизм каталитического акта является развитием представлений Лорана и Кекуле, а также некоторых физико-химиков начала XX века (Марселин) о промежуточной конфигурации атомов в процессе превращения. Баландиным была отмечена необходимость структурного соответствия между строением реагирующих молекул и кристаллической решеткой катализатора для того, чтобы произошла хемосорбция. Мультиплетная теория посредством принципа структурного соответствия ввела матричный эффект в катализ и хорошо объяснила многие случаи ориентации реакций, т.е. их селективной направленности. Однако энергетическая часть мультиплетной теории не давала химикам принципиальной возможности неэмпирического расчета энергетических характеристик системы. Только после разработки теории абсолютных скоростей была создана принципиальная схема расчета скоростей реакций.
Цепная теория катализа.
В 1930 годах было показано, что стенки участвуют и в зарождении цепей. В 1940-х годах М.В, Поляков доказал, что почти все цепные реакции по существу представляют гетерогенно-гомогенные каталитические превращения.
В 1950-х годах В,В, Воеводский, Ф.Ф. Волькенштейн и Н.Н. Семенов ввели в цепную теорию принципиально новый момент — непрерывность образования свободного радикала на стенке или вообще на твердом катализаторе.
Химическая концепция активной поверхности.
В 20-30-х годах в трудах С.З. Рогинского явилось учение об активной поверхности, Рогинский пришел к выводу о зависимости каталитической активности твердого тела от его электронного строения, Впоследствии, показав, что каталитическая активность связана с проводниковыми свойствами. Рогинский подразделил катализ на два класса: электродный (окислительно-восстановит>ельный) и ионный (кислотно-основной).
В связи с теорией гетерогенного катализа в 30-40-х годах усиленно разрабатывали, в особенности в Америке, вопрос о природе катализирующих поверхностей (теория Тейлора). Теория Тейлора — это воззрение на особое строение поверхности катализатора, теория, которая связывает адсорбцию с каталитическим действием. Такое представление о роли катализатора ведет к дальнейшему проникновению в понимание гетерогенных каталитических реакций, которые в каталитическом поле идут в пограничных областях двух: фаз.
Электронная теория.
Работы Рогинского послужили отправным пунктом для создания электронной теории катализа, имеющей ряд направлений развития. Наиболее общие из этих направлений — теория полупроводникового катализа. Эта теория устанавливает зависимость каталитической активности от количества захваченных или специально введенных в кристалл примесей и от природы этих примесей (доноры, акцепторы электронов), а также от формы и размеров первичных кристаллов, от внешних факторов (действие света, подогрев), от электрического поля, окружающего проводник.
Она до сих пор подробно развита лишь в применении к окислительно-восстановите>льным реакциям.
Теория кислотно-основного катализа.
В 1920-х годах Бренстед и Лоури интерпретировали кислотно-основной катализ как протолитическую реакцию между субстратом (слабой кислотой или основанием) и катализатором (сильным основанием или кислотой):
В 1940-х годах М. Девис, НА. Измайлов и другие химики показали более сложный характер механизма диссоциации кислот и оснований. При этом переход протона представляет лишь конечную и не всегда обязательную стадию взаимодействия. Главной же формой кислотно-основного катализа является образование молекулярных соединений, в которых водородная связь есть форма проявления (и в ряде случаев незавершенности) протонного перехода:
В настоящее время теория кислотно-основного катализа применяется главным образом для гомогенных реакций. Для рассмотрения с единой точки зрения как гомогенных, так и гетерогенных каталитических превращений большое значение имеют теории позволяющие количественно учитывать активные центры катализатора. Именно такую теорию (активных ансамблей) выдвинул Н.И, Кобозев в 1939 году.
Теория активных ансамблей.
Теория аггравации.
Согласно этой теории, носителем каталитической активности являются не кристаллические тела а группы атомов (ансамбли), располагающиеся на «инертной подкладке», на собственной кристаллической решетке или находящиеся в виде паров металла в объеме.
Интересны также идеи Кобозева об аггравации —- захвате энергии реакции той частью катализатора, которая лишь несет активный центр, представляя утяжелитель агграватор. Агграватор выполняет роль энергетической ловушки. Полученная таким образом энергия идет на возбуждение активного центра, активность которого по мере усложнения агграваторов возрастает по экспоненциальному закону. Сущность и механизм этого вида активации еще не ясны. Но вопрос об эффективности аггравации в общей проблеме катализа приобретает очень важное значение.
В 40-х годах выдвигается вопрос о значении и преимуществе смешанных катализаторов, которые ускоряют реакцию и направляют ее в одну определенную сторону, Митташ показал, что оксид железа как катализатор, вследствие примеси оксида висмута, настолько хорошо катализирует окисление аммиака, что такой контакт может практически заменить платину.
Смесь катализаторов иначе реагирует, чем сами они отдельно взятые, и в смесях индивидуальные химические свойства элементов в значительной степени подавляются, появляются новые свойства смеси, которая реагирует как самостоятельное вещество.
Из работ по катализу, проведенных в 40-50-х годах XX века можно сделать следующие выводы:
1. Прежде всего, катализатор должен удовлетворять определенным объективным признакам. Из изученных процессов дегидрогенизации гидрогенизации видно, что только те металлы являются хорошими катализаторами, радиусами которых находятся в пределах 1,236-1,397 .
2. Адсорбция при катализе не играет доминирующей роли, хотя катализ и связан с адсорбцией. Отравленный катализатор ведет реакцию по другому пути, отчего и возникают побочные реакции конденсации при катализе органических соединений.
3. Для того чтобы катализатор работал, необходимо создать подходящие для него термодинамические условия. Они не одинаковы для разных катализаторов.
4. Температурный коэффициент скорости реакции не зависит от природы катализируемого вещества для данного типа реакции, а только от природы примененного катализатора.
Заключение
Идеи Берцелиуса и Либиха так и остались гениальными предвидениями, не опирающимися на надежный фундамент представлений о динамике процесса. Только после разработки первых теорий формальной кинетики в 80-х годах XIX века начали интенсивно развиваться теории катализа (контактные представления Вант-Гоффа и Оствальда). Новые успехи в развитии кинетики в XX веке (коллизионные представления, теории разветвленных цепей реакций и абсолютных скоростей реакций) порождали дальнейшее совершенствование представлений о катализе в то время: теория промежуточной хемосорбции, цепная и электронная теория катализа химическая концепция активной поверхности, теория кислотно-основного катализа и оксидных ансамблей. Специфика каталитических теорий обусловлена как самим характером соотнесения кинетики и катализа, так и важным промышленным значением последнего. Недаром объектами кинетического исследования оказывались главным образом хорошо известные химикам и производственникам каталитические превращения (например, основной постулат химической кинетики был сформулирован Вильгельми при изучении инверсии тростникового сахара, а затем подтвержден Бертло при исследовании омыления и этерификации). Тесное историческое и логическое переплетение проблем кинетики и катализа обусловило и общую задачу, стоящую перед этим динамическим направлением химии — создание фундаментальной количественной теории протекания реакций.
Одной из интересных попыток решения этой проблемы (наряду с развитием квантово-механических исследований и корреляционного анализа) явилось создание теории саморазвития электронных каталитических систем. основанное на последовательном применении динамического метопа, предполагающего переменность всех основных параметров катализа (природа катализатора, постоянство активности и специфичности катализатора и механизм каталитического акта).
В настоящее время работы по катализу вступили в новую фазу своего развития. Наметилось заметное сближения между гомогенно-гетерогенным и ферментативным видами катализа, Химики все чаще обращаются к ферментативному катализу не только как к объекту исследования, но и как к источнику новых идей способных помочь в создании более эффективных химических процессов.
Создание новых высокоэффективных катализаторов, по-видимому, может быть осуществлено в результате использования принципов структурной организации ферментов, в присутствии которых реакции протекают со значительно большими скоростями, чем в присутствии синтетических катализаторов и в весьма мягких условиях
|




